





Abstract
Summary: The semantic comparisons of Gene Ontology (GO) annotations provide quantitative ways to compute similarities between genes and gene groups, and have became important basis for many bioinformatics analysis approaches. GOSemSim is an R package for semantic similarity computation among GO terms, sets of GO terms, gene products and gene clusters. Four information content (IC)- and a graph-based methods are implemented in the GOSemSim package, multiple species including human, rat, mouse, fly and yeast are also supported. The functions provided by the GOSemSim offer flexibility for applications, and can be easily integrated into high-throughput analysis pipelines.
Availability: GOSemSim is released under the GNU General Public License within Bioconductor project, and freely available at http://bioconductor.org/packages/2.6/bioc/html/GOSemSim.html
Contact: boxc@bmi.ac.cn; sqwang@bmi.ac.cn
Supplementary information: Supplementary information is available at Bioinformatics online.
1 INTRODUCTION
The Gene Ontology (GO) is becoming the de facto standard for the annotation of gene products. The GO consortium annotates gene products with terms from three orthogonal ontologies organized as directed acyclic graphs, laying the foundation for quantitative semantic comparisons. Several metrics have been proposed to measure the semantic similarity between GO annotations, and have been verified in terms of the correlations with sequence similarity (Lord et al., 2003) protein–protein interactions (Xu et al., 2008), and gene expression profiles (Sevilla et al., 2005). The GO semantic similarity provides the basis for functional comparison of gene products, and therefore has been widely used in bioinformatics applications, such as protein sub-nuclear localization prediction (Lei and Dai, 2006), gene function prediction (Tao et al., 2007) and cluster analysis of genes (Bolshakova et al., 2005; Wolting et al., 2006).
Two typical approaches to measure semantic similarity of GO terms are information content (IC)- and graph-based measures. The IC-based measures depend on the frequencies of two GO terms involved and that of their closest common ancestor term in a specific corpus of GO annotations, such as the UniProt Knowledgebase. Three IC-based measures, Resnik's (Philip, 1999), Lin's (Lin, 1998) and Jiang and Conrath's (Jiang and Conrath, 1997) have been introduced from natural language taxonomies by Lord et al. (2003) to compare gene products in early time. On the basis of Resnik's and Lin's definition, an IC-based measure has also been presented by Schlicker et al. (2006). Considering that the specificity of a GO term is usually determined by its location in the GO graph, Wang et al. (2007) proposed a graph-based strategy to compute semantic similarity using the topology of the GO graph structure. In the Wang's method, the semantics of GO terms are encoded into a numeric format and the different semantic contributions of the distinct relations are considered.
Several online tools for semantic similarity measurement of gene products are available at present, such as G-SESAME (Wang et al., 2007) and FuSSiMeG (http://xldb.fc.ul.pt/rebil/ssm/). To facilitate large-scale analysis, two freely available software packages, GOGraph (Lord et al., 2003) and GOSim (Frohlich et al., 2007) implementing classic IC-based methods for semantic comparison of GO terms have also been developed. Here, we present an R package named GOSemSim to compute semantic similarity among GO terms, sets of GO terms, gene products and gene clusters, providing both IC- and graph-based methods.
2 IMPLEMENTATION
The GOSemSim is developed as a package for the statistical computing environment R and is released under the GNU General Public License within Bioconductor (Gentleman et al., 2004) project. GOSemSim depends on the annotation data GO.db provided by Bioconductor to obtain the ancestors of GO terms and their relations. The information content is species specific and calculated from Bioconductor annotation packages org.Hs.eg.db, org.Rn.eg.db, org.Mm.eg.db, org.Dm.eg.db and org.Sc.sgd.db for human, rat, mouse, fly and yeast, respectively.
Considering that existing approaches performs differently under different circumstances (Pesquita et al., 2009), four IC-based (Resnik's, Lin's, Jiang and Conrath's and Schlicker's) and one graph-based (Wang's) semantic similarity measure algorithms mentioned before are selected to be integrated in GOSemSim, and can be selected by setting the ‘method’ parameter of the package functions to ‘Resnik’, ‘Lin’, ‘Jiang’, ‘Rel’ and ‘Wang’, respectively. The Resnik's, Lin's and Jiang and Conrath's algorithms are the most common semantic similarity measures used with GO (Pesquita et al., 2009). Several assessment results had shown that the Resnik's measure had consistently high correlation with sequence similarity (Lord et al., 2003; Mistry and Pavlidis, 2008; Pesquita et al., 2008) and gene co-expression (Sevilla et al., 2005). By using a best-match average combination strategy, Pesquita et al. found that Jiang and Conrath's measure had the highest correlation with sequence similarity (Pesquita et al., 2009). The Schlicker's measure had been found to perform better than Resnik's measure in distinguishing orthologous gene products from gene products with other levels of sequence similarity (Schlicker et al., 2006). The Wang's measure had also been shown to produce more accurate results than Resnik's measure in clustering gene pairs according to their semantic similarity (Wang et al., 2007). The details about the semantic similarity measure algorithms used in GOSemSim can be found in the user's manual (Supplementary Material 1). The GO used in measurement can be restricted by assigning the corresponding parameter to ‘BP’ (biological process), ‘MF’ (molecular function) and ‘CC’ (cellular component).
3 FUNCTIONS AND EXAMPLES
Six functions are provided by GOSemSim package. The function goSim, mgoSim, geneSim and clusterSim can compute the semantic similarity among GO terms, sets of GO terms, GO descriptions of gene products and GO descriptions of gene clusters, respectively. The functions mgeneSim and mclusterSim are designed to calculate the similarity scores matrix of a set of genes and gene clusters.
The output value of the basic function goSim is between 0 and 1. The higher the value obtained more the similarity between them. For example:
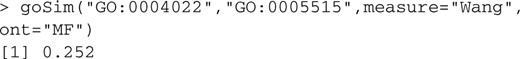
The function mgoSim is designed to compute the similarity of two GO terms lists, such as

By mapping gene products to GO annotations, functions geneSim, mgeneSim, clusterSim and mclusterSim can be used to measure the semantic similarity among gene products. Gene IDs and species are needed for the measurements. For human, rat, mouse and fly, the Gene IDs refer to Entrez Gene IDs, while for yeast, the Gene IDs refer to ORF identifiers from Saccharomyces Genome Database (SGD), for example:

The functions mgeneSim, clusterSim and mclusterSim are especially designed for large-scale analysis. Suppose we have a group of genes (here, we use a random sample set of Affymetrix IDs as an example) and want to cluster the genes based on their functions. First, we call the function mgeneSim to compute the pairwise GO semantic similarities of these genes:

Then, we can use hierarchical cluster function hclust of the stats package to cluster these gene products based on semantic similarities of their GO annotations. After cutting the cluster tree into discrete cluster groups, we can use clusterSim and mclusterSim function to measure the similarities among gene clusters, for instance:

4 CONCLUSIONS
The measurements of the semantic similarities for GO annotations facilitate users to infer relationships among genes, and therefore is becoming one of the important bases in many bioinformatics analysis methods. The GOSemSim package implement five classic approaches for GO annotations-based semantic similarity measurements, and provide useful functions to offer flexibility for typical applications. The package can be easily integrated into pipelines for high-throughput analysis, such as gene expression data mining, protein interactions validation and miRNA-regulated network interpretation.
Funding: National Key Technologies R&D Program for New Drugs (2009ZX09301-002); National Nature Science Foundation of China (30530650); National Science Fund for Distinguished Young Scholars (30625041).
Conflict of Interest: none declared.
REFERENCES
Author notes
†The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.
Associate Editor: Olga Troyanskaya

