





Abstract
Motivation: Linking gene mentions in an article to entries of biological databases can facilitate indexing and querying biological literature greatly. Due to the high ambiguity of gene names, this task is particularly challenging. Manual annotation for this task is cost expensive, time consuming and labor intensive. Therefore, providing assistive tools to facilitate the task is of high value.
Results: We developed GeneTUKit, a document-level gene normalization software for full-text articles. This software employs both local context surrounding gene mentions and global context from the whole full-text document. It can normalize genes of different species simultaneously. When participating in BioCreAtIvE III, the system obtained good results among 37 runs: the system was ranked first, fourth and seventh in terms of TAP-20, TAP-10 and TAP-5, respectively on the 507 full-text test articles.
Availability and implementation: The software is available at http://www.qanswers.net/GeneTUKit/.
Contact: aihuang@tsinghua.edu.cn
1 INTRODUCTION
Gene normalization is one of the most challenging tasks in bio-literature mining due to the high ambiguity of gene names as they may refer to orthologous or entirely different genes, may be named after phenotypes and other biomedical terms, or may resemble common names with non-gene entities (Hakenberg et al., 2008). It is time consuming and labor intensive to annotate full-text articles manually. Therefore, a good assistive tool for this task may facilitate the process greatly.
There has been a large body of work addressing the problem of gene mention normalization. ProMiner (Hanisch et al., 2005), a strict dictionary-based approach, relies on the quality of its gene dictionaries heavily. Xu et al. (2007) proposed a method using gene profiles generated from PubMed abstracts for gene disambiguation. GNAT (Hakenberg et al., 2008) is a rule-based and machine learning (ML) based gene normalization system which used extensive background knowledge. Built from open-source libraries and publicly available resources, GENO (Wermter et al., 2009) employed a carefully crafted suite of symbolic and statistical methods. Moara (Neves et al., 2010) is a Java library for extracting and normalizing gene and protein mentions, and currently designed for four model organisms.
Our software departs from previous systems in two aspects: first, it combines local and global contexts to normalize genes at document-level. The goal of this software is not to normalize every mention correctly, but to suggest a list of normalized genes given a target document, to assist human annotators. Most previous systems are normalizing genes at mention-level and only local context surrounding a mention (e.g. the sentence where the mention was recognized) were employed. However, due to the high ambiguity of gene names, it may be insufficient to use only local context: inter-sentential or document-level context can be helpful in this task. Second, the software is designed for simultaneously normalizing genes of many different species for full-text articles. It is not limited to any specific organism, but rather deals with all species present in a gene database (Entrez Gene in this article).
2 METHODS AND SYSTEM
The workflow of our software is shown in Figure 1. The software has four main modules. The first module is for gene mention recognition, the second one for gene ID candidate generation and the third one for gene ID disambiguation. In the fourth module, the software generates confidence scores for each gene ID, where the confidence score indicates the strength of the association between a gene ID and the document.
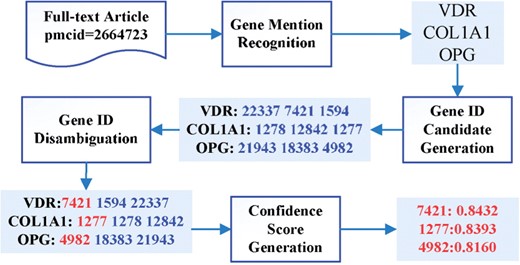
The workflow of GeneTUKit. Numbers in shaded boxes are gene IDs. The real-number values in the last box are confidence scores.
We have used three methods for recognizing gene mentions in the first module. The first method is a conditional random field-based approach, which was trained on the training dataset of BioCreAtIvE II Gene Mention Recognition Task (Smith et al., 2008). The second method is a dictionary-based recognition approach where the dictionary was compiled from Entrez Gene. The third method is ABNER (Settles, 2005), an open source named entity recognition system for biomedical literature. The input text is processed by these methods separately, and the resulting mentions are maintained if a mention is recognized by at least two methods. If two mentions are similar but have different boundaries, the overlapping part is taken as the final mention.
The second module generates gene ID candidates for a recognized mention. In this module, an open-source indexing package, Lucene (http://lucene.apache.org/), was used to index all the genes in Entrez Gene. Each mention was then queried and top 50 gene IDs were returned as candidates. The text of mentions and Entrez Gene entries were, respectively, processed by the following rules sequentially: (i) removing special characters such as dashes and underscores; (ii) removing stop words; (iii) changing words such as ‘hBCL’ into ‘h BCL’; (iv) separating digits, Greek and Roman letters from alphabetic letters; and (v) converting the text to lowercase letters.
The third module is for disambiguating gene IDs, which is accomplished by a ranking algorithm. The algorithm was trained on the 32 full-text articles provided by BioCreAtIvE III. Each article has a list of tuples (gene mention, gene id and species); however, the annotations did not give the positions where a gene mention was recognized. The training samples were generated as follows: for each gene ID candidate, if the ID appears in the manual annotation list, the candidate is taken as positive, otherwise negative. For each gene ID candidate and its corresponding mention, we extract features from local and global contexts. Some local context features are as follows: The document-level, global context features are listed partly as follows: In constructing these features, we used dictionary-based matching to recognize species as such a simple method can produce fairly good performance. For finding full/abbreviated name mappings, we adopted a method from (Schwartz and Hearst, 2003). Once features were obtained, we used a ranking algorithm ListNet (Cao et al., 2007) to rank gene IDs for each mention and the top one ID was maintained for further processing.
The ranking score of the gene ID given by the Lucene index.
Whether the species of the ID is implied by the gene mention, such as hBCL.
The edit distance between the mention and the official symbol of the ID.
The minimal edit distance between the mention and all synonyms of the ID.
Whether at least one word indicating gene functions of a gene ID appears in the sentences from which the mention was recognized. The words indicating gene functions are obtained from the corresponding gene symbols after removing common words (such as protein, gene etc.) and words containing capital letters or digits (e.g. VDR, p65).
Whether the species of the gene ID appears in the document.
Whether the species of the ID appears in the title.
Whether the species of the ID is the nearest species in the same paragraph where the mention is recognized.
If the mention has a full (or abbreviated) name through the document, compute the minimal edit distance between synonyms of the ID and the full (or abbreviated) name of the mention.
The fourth module generates a confidence score for each predicted gene ID to measure the association of the given gene ID and the document using a support vector machine (SVM) classifier. The training examples were constructed similarly as in the third module. The features were constructed as follows:
The best value of features used in the third module as each ID may correspond to many mentions. For the edit distance features, ‘best’ means ‘minimal’; for the ranking score feature, ‘best’ means ‘maximal’.
The total number of gene mentions associated with the ID.
The highest rank of the ID among all the mentions associating with the ID.
3 RESULTS
We evaluated the system on the BioCreAtIvE III GN corpus (Lu and Wilbur, 2010) in terms of Threshold Average Precision (TAP-k, k = 5, 10, 20, respectively) (Carroll et al., 2010). For training, we used the 32 articles with gold-standard human annotation. For testing, the first dataset has 50 articles, each of which has gold-standard annotation, and the second one has 507 articles whose ground truth was inferred from 37 team submissions (referred as silver standard). The 507 articles also include the 50 articles from the first dataset. The results presented in Table 1 show the official evaluation results from BioCreAtIvE III. We have also tested the performance in terms of average precision. The manual error analysis has revealed that two major error types are (i) wrongly recognized gene mentions, and (ii) wrong species mapping. The Supplementary Material provide a more detailed analysis at http://www.qanswers.net/GeneTUKit/evaluation.html.
The evaluation results on the BioCreAtIvE III GN corpus
| Measures | 50 articles (gold standard) | 507 articles (silver standard) |
|---|---|---|
| TAP-5 | 0.2973 (4/37) | 0.4086 (7/37) |
| TAP-10 | 0.3125 (4/37) | 0.4511 (4/37) |
| TAP-20 | 0.3248 (4/37) | 0.4648 (1/37) |
| Average precision of TOP k recommendations | ||
| k = 5 | 0.4880 | 0.5764 |
| k = 10 | 0.4340 | 0.4993 |
| k = 20 | 0.3231 | 0.3984 |
| Measures | 50 articles (gold standard) | 507 articles (silver standard) |
|---|---|---|
| TAP-5 | 0.2973 (4/37) | 0.4086 (7/37) |
| TAP-10 | 0.3125 (4/37) | 0.4511 (4/37) |
| TAP-20 | 0.3248 (4/37) | 0.4648 (1/37) |
| Average precision of TOP k recommendations | ||
| k = 5 | 0.4880 | 0.5764 |
| k = 10 | 0.4340 | 0.4993 |
| k = 20 | 0.3231 | 0.3984 |
The number in the bracket is the rank of our score among the 37 submissions.
The evaluation results on the BioCreAtIvE III GN corpus
| Measures | 50 articles (gold standard) | 507 articles (silver standard) |
|---|---|---|
| TAP-5 | 0.2973 (4/37) | 0.4086 (7/37) |
| TAP-10 | 0.3125 (4/37) | 0.4511 (4/37) |
| TAP-20 | 0.3248 (4/37) | 0.4648 (1/37) |
| Average precision of TOP k recommendations | ||
| k = 5 | 0.4880 | 0.5764 |
| k = 10 | 0.4340 | 0.4993 |
| k = 20 | 0.3231 | 0.3984 |
| Measures | 50 articles (gold standard) | 507 articles (silver standard) |
|---|---|---|
| TAP-5 | 0.2973 (4/37) | 0.4086 (7/37) |
| TAP-10 | 0.3125 (4/37) | 0.4511 (4/37) |
| TAP-20 | 0.3248 (4/37) | 0.4648 (1/37) |
| Average precision of TOP k recommendations | ||
| k = 5 | 0.4880 | 0.5764 |
| k = 10 | 0.4340 | 0.4993 |
| k = 20 | 0.3231 | 0.3984 |
The number in the bracket is the rank of our score among the 37 submissions.
4 CONCLUSION
GeneTUKit is a software designed for document-level gene normalization, which employs features from the local context and the global context within the whole full-text article. It can normalize genes of many different species. Given a target article, the software outputs a list of normalized genes, and each predicted gene is associated with a confidence score.
Funding: Natural Science Foundation of China (No. 60803075); Chinese 973 project (No. 2007CB311003).
Conflicts of Interest: none declared.
REFERENCES
Author notes
Associate Editor: Jonathan Wren


{kind=link}