





Abstract
Summary: There is increasing evidence showing that co-expression of genes that cluster along the genome is a common characteristic of eukaryotic transcriptomes. Several algorithms have been used to date in the identification of these kinds of gene organization. Here, we present a web tool called CROC that aims to help in the identification and analysis of genomic gene clusters. This method has been successfully used before in the identification of chromosomal clusters in different eukaryotic species.
Availability: The web server is freely available to non-commercial users at the following address: http://metagenomics.uv.es/CROC/
Contact: miguel.pignatelli@uv.es
1 INTRODUCTION
It is well documented that high-order organization of genes occurs in the chromosomes of many eukaryotic genomes like yeast, worm, fly, mouse and human [see Hurst et al. (2004) for a comprehensive review]. In some cases, these genes appear to be functionally related (Blanco et al., 2008), evolutionarily conserved (de Wit et al., 2008; Poyatos and Hurst, 2007) or even belong to the same protein–protein interaction network (Teichmann and Veitia, 2004). A recent study determined that most of the transcription factors in Saccharomyces cerevisiae tend to bind targets that are positionally clustered within a specific region on the chromosome (Janga et al., 2008). Different mechanisms such as gene duplication, shared promoter regions, chromatin regulation or shared functional pathway or tissue have been proposed to explain the existence of these clusters (Hurst et al., 2004; Lercher et al., 2003; Oliver et al., 2002).
To date, different statistical models have been used to assess the significance of physical clusters. Recently, a strategy consisting of serial hypergeometric distribution tests in a sliding window along the chromosomes has been successfully used in some studies (Chang et al., 2004; Coppe et al., 2006; Yi et al., 2007). Following this approach, Coppe et al. developed a software called REEF that implements this algorithm in a python program. Using this software, they were able to identify 44 significant tissue-specific clusters in the human genome (Coppe et al., 2006). Using a similar approach, Yi et al. (2007) found gene clusters that share a common Gene Ontology (GO) annotation term in different genomes.
Here we present CROC, a web-based tool for the identification of chromosomal clustering in a wide variety of eukaryotic genomes. It uses a sliding window approach combined with hypergeometric distribution tests to evaluate the statistical significance of the predicted clusters. The most important parameters of the algorithm are customizable through a simple and powerful web interface. The results are linked to external databases for further analysis. Recently, using this algorithm we were able to identify different conserved clusters governed by chromatin regulators in Drosophila melanogaster (Blanco et al., 2008).
2 IMPLEMENTATION
The core of the CROC application has been implemented in Perl. The statistical analysis routines are written in C to maximize efficiency and inlined directly in the Perl code. All scripts are available for download in the web server page. The web interface uses Javascript routines heavily to maximize user friendliness. The communication between the web interface and the application's core is handled via AJAX techniques and JSON has been used for information exchange between both.
2.1 Data input
The program expects a list of genes, for instance a list of genes up- or down-regulated after a microarray experiment. These are the genes that will be tested for physical clustering. Only valid IDs for the selected genome/version are allowed, for example, for UCSC genomes, gene abbreviations (e.g. human ‘BCL2’ or ‘LDHAL6B’) or refseq ID's (e.g. NM_198576) are both allowed. Also, the reference genome and its version need to be selected. Examples of data input for different organisms can be obtained ready for analysis in the ‘Examples’ box. It is also possible to upload your own custom reference.
2.2 Other options
Three other parameters that control the behavior of the algorithm can be adjusted. The first one is the window type that the algorithm will use in the sliding-window strategy. Two types of windows can be selected, gene-based and DNA length-based. In the first case, the offset between windows will always be one gene. In the latter, the offset can be selected between 1 kb and the length of the window. The second parameter refers to the minimum number of input genes that define a cluster. Finally, the method for multiple testing correction can be selected.
2.3 Algorithm
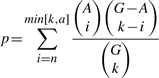
2.4 Output
The output is distributed in three sections. The first one (‘Stats’) shows a brief summary of the analysis performed. The second section (‘Chromosomes’) summarizes the results disclosed by chromosomes. A graphical representation of the chromosome can be generated by clicking the ‘Plot’ button in the last field of each chromosome row. In this plot, gray lines represent all the genes present in the chromosome, red lines represent input and the blue lines over the genes represent clusters. Because these blue lines are typically very short, blue arrows are present over them to ensure visibility of all clusters. When available genomes are used, these arrows are themselves link to the UCSC/Ensembl genome browser, where clusters are uploaded as a ‘custom track’ to further analyze them. The third section (‘Clusters’) depicts individual information for each cluster including: the chromosomal coordinates of the cluster, the associated P-value of the statistical analysis, the input genes that form the cluster and a plot representing the cluster itself.
3 CONCLUSION
The tool presented here aims at helping in the identification of gene clusters in eukaryotic chromosomes. The input list can represent genes that share any biological feature, such as, for example, co-expression, having a common transcription factor in their proximal promoter, belonging to the same GO term or being fast-evolving genes. We have recently demonstrated the usefulness of this algorithm in studying clustered genes governed by chromatin regulators during the development of D. melanogaster (Blanco et al., 2008).
ACKNOWLEDGEMENTS
We thank Ana Pamblanco for help in the web page design and Juanjo Abellán for his helpful comments. MP was supported by Juan de la Cierva postdoctoral contract from the Ministerio de Educación y Ciencia (MEC), Spain.
Funding: This work was supported by the Ministerio de Educación y Ciencia, Spain [BMC2003-05018,BMC2006-07334].
Conflict of Interest: none declared.
REFERENCES
Author notes
Associate Editor: Dmitrij Frishman

